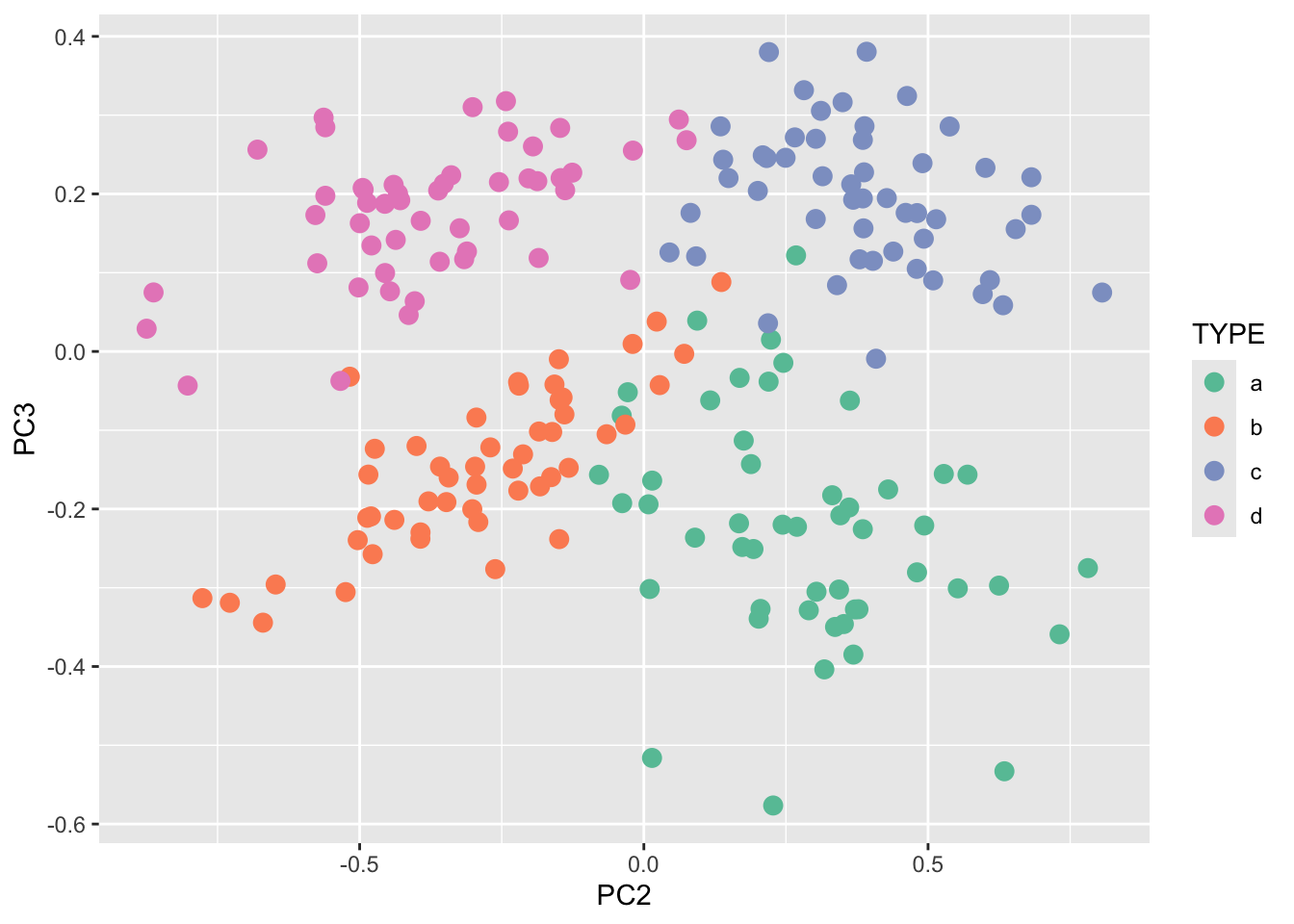
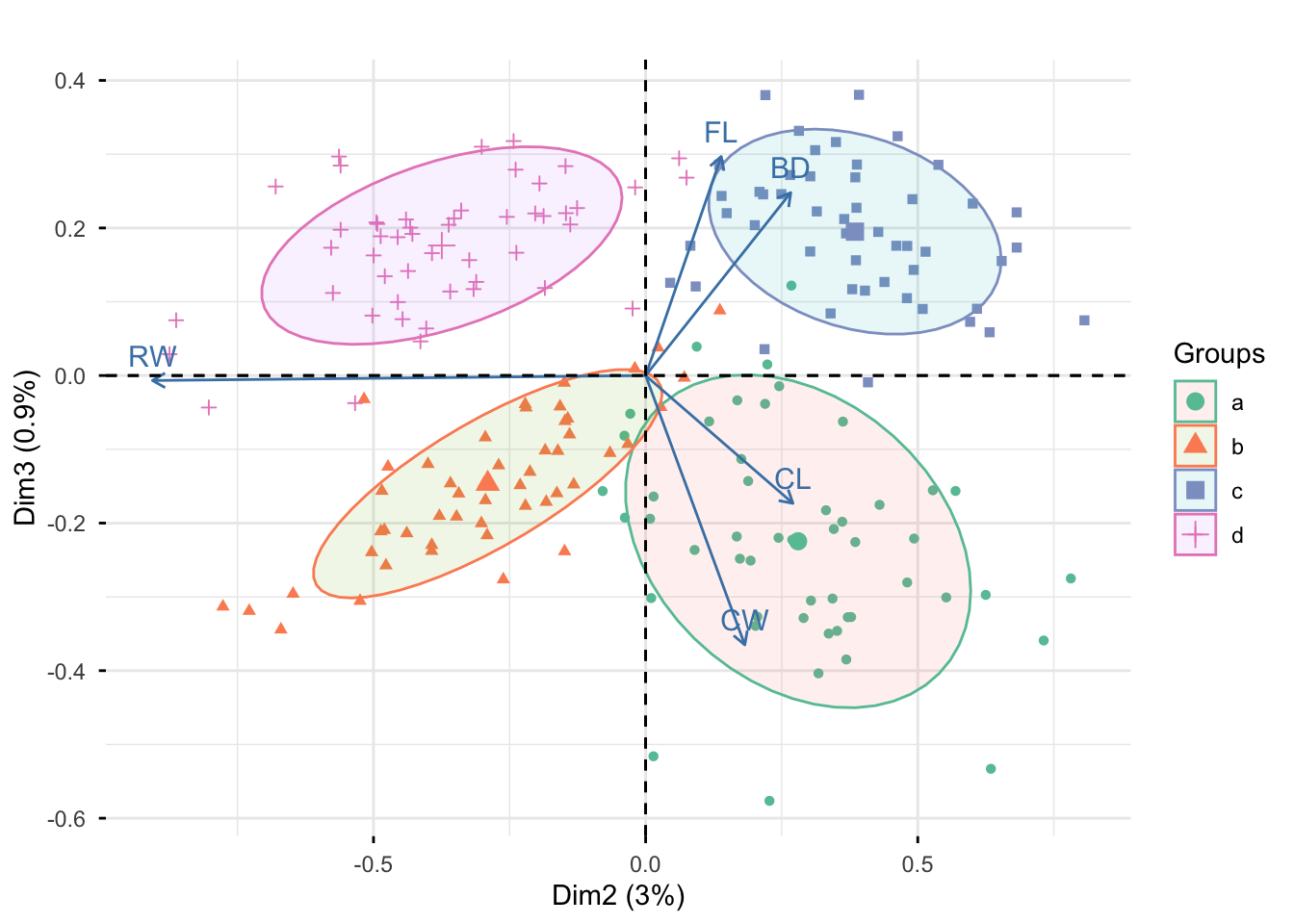
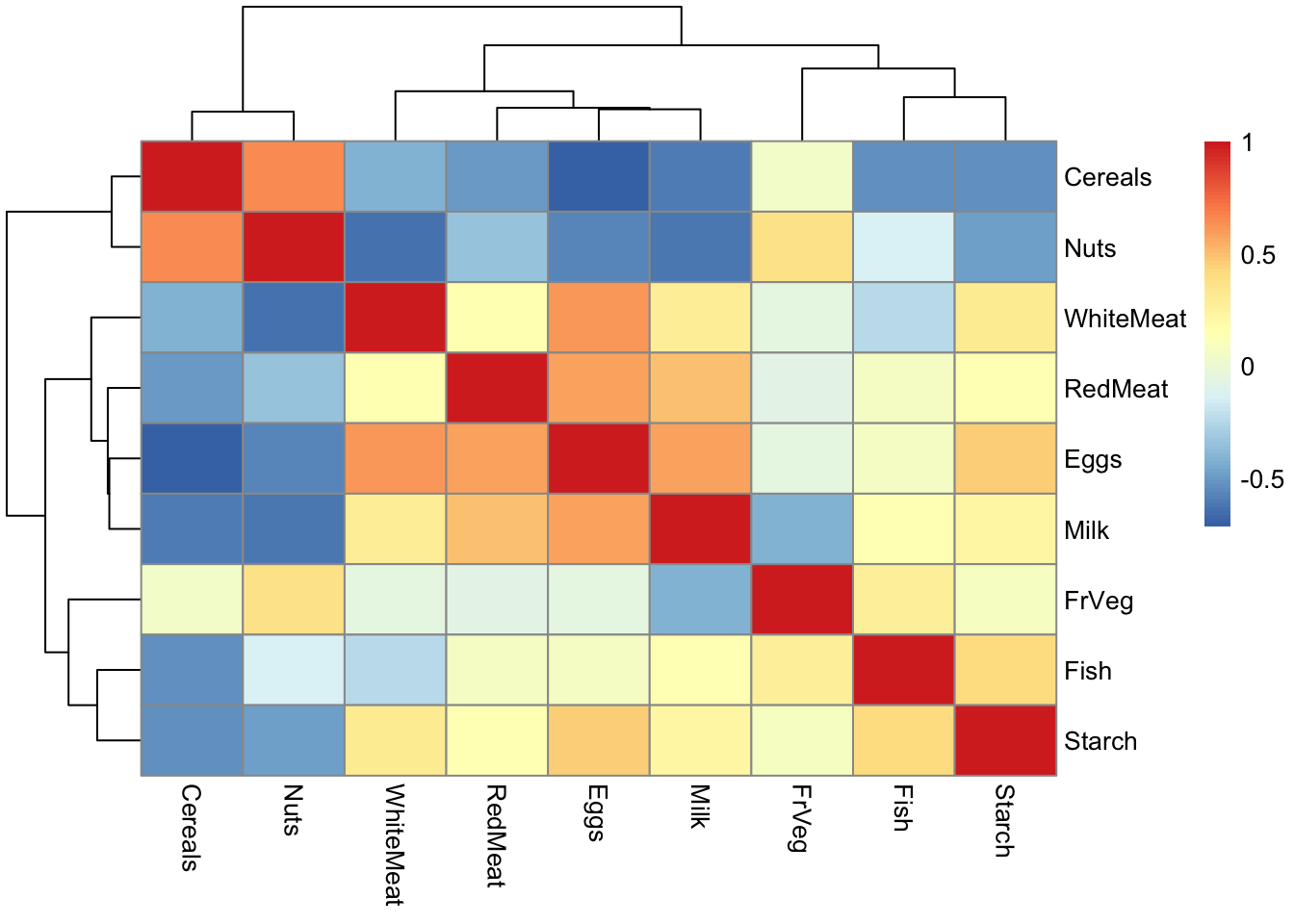
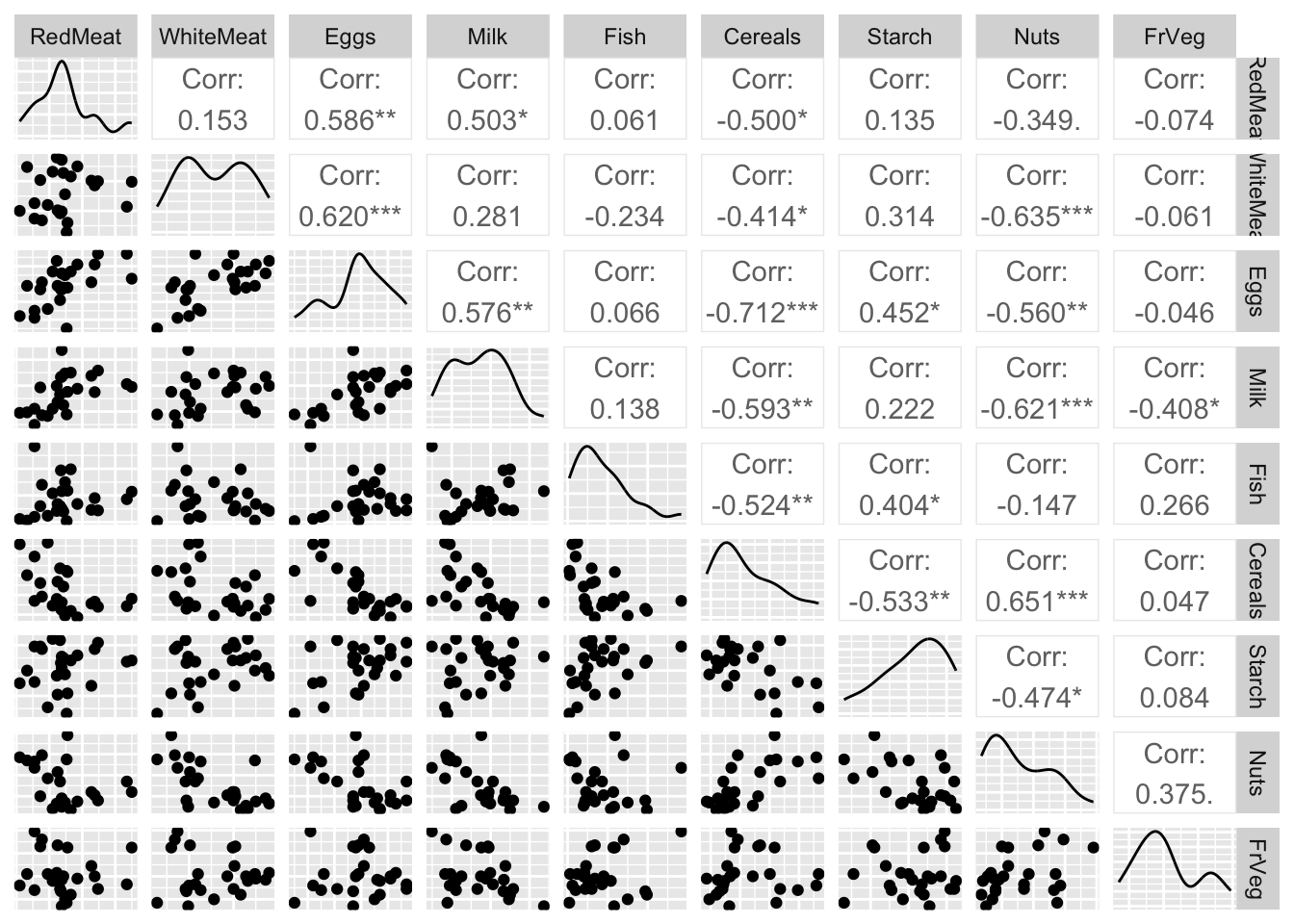
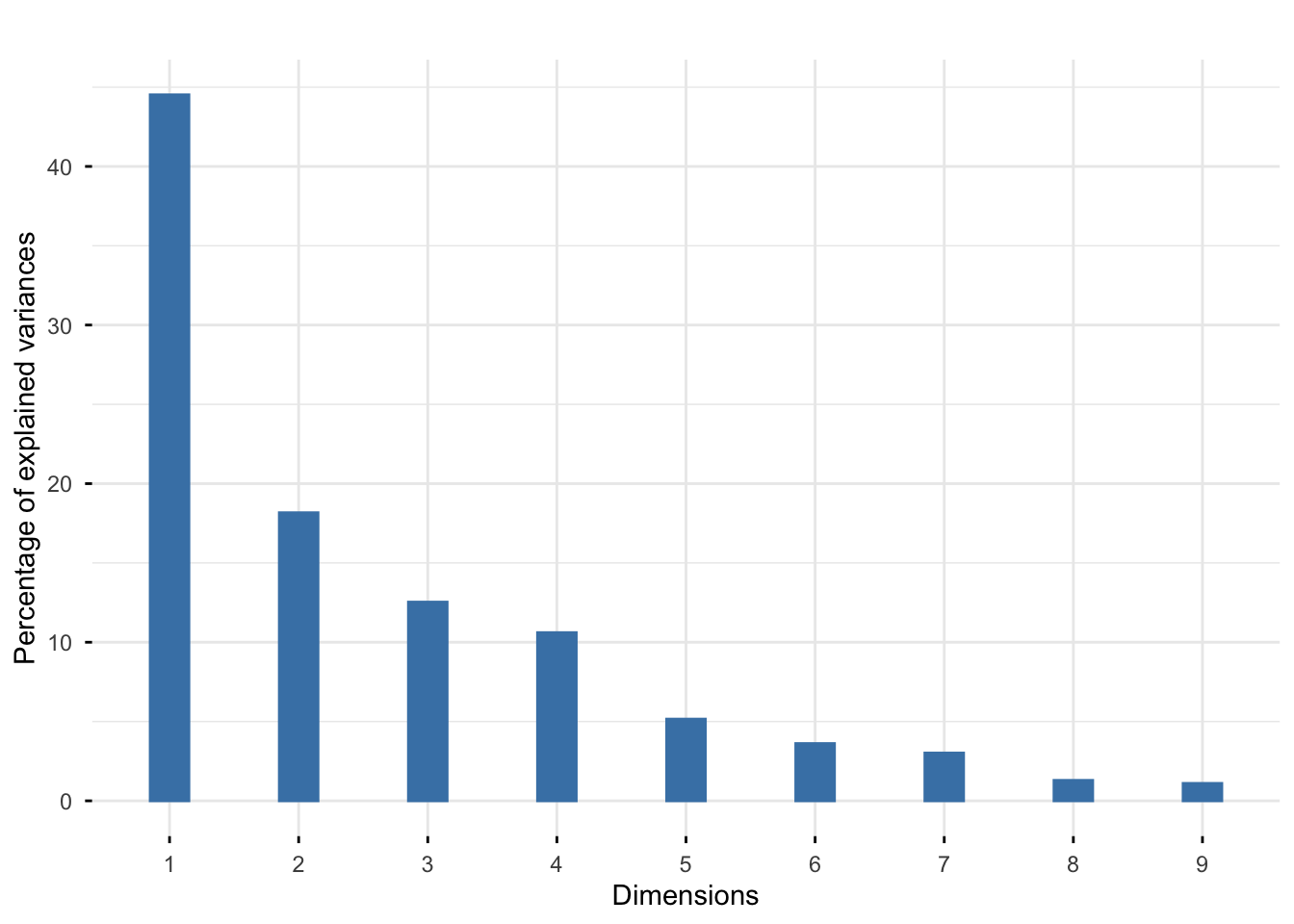
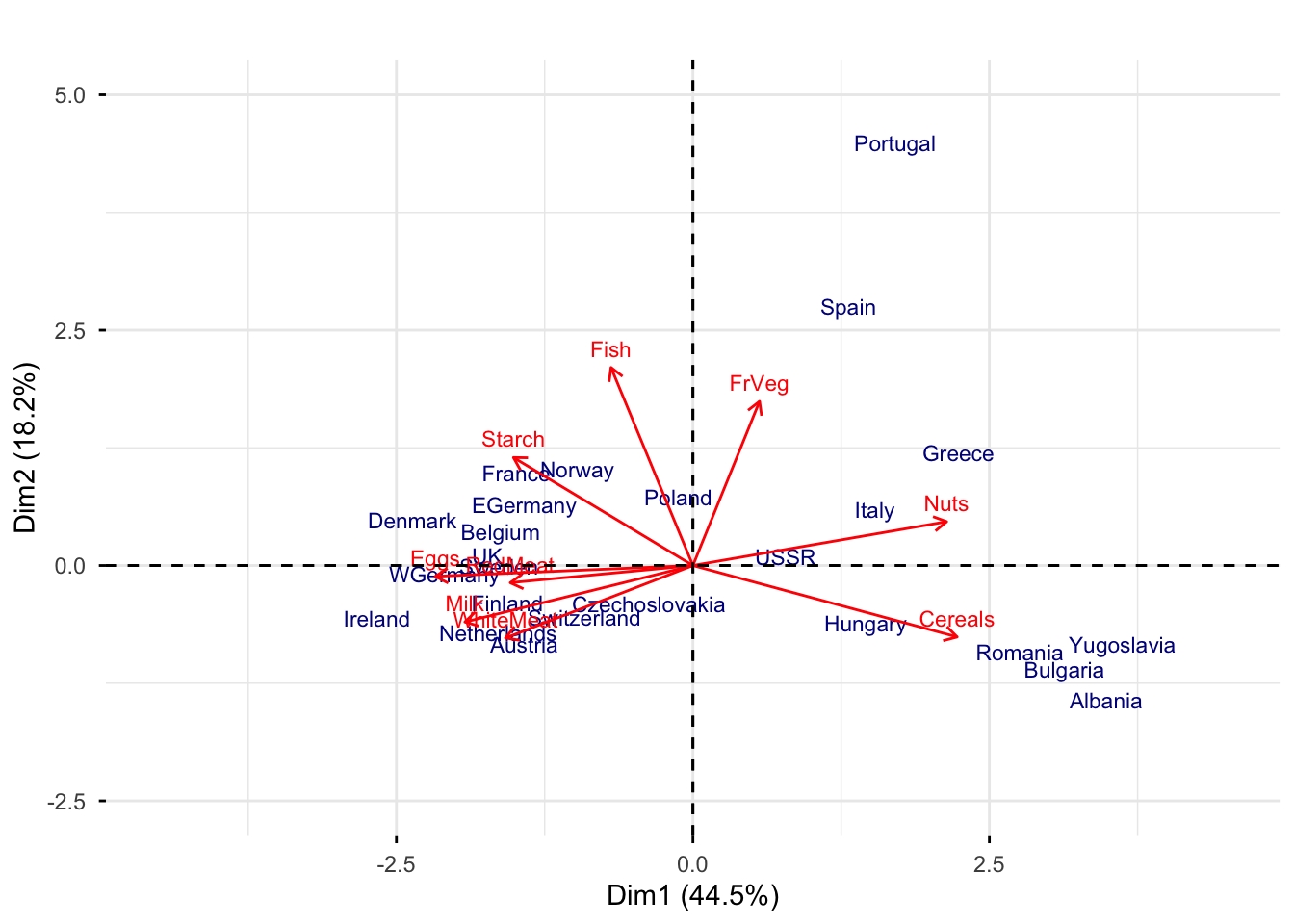
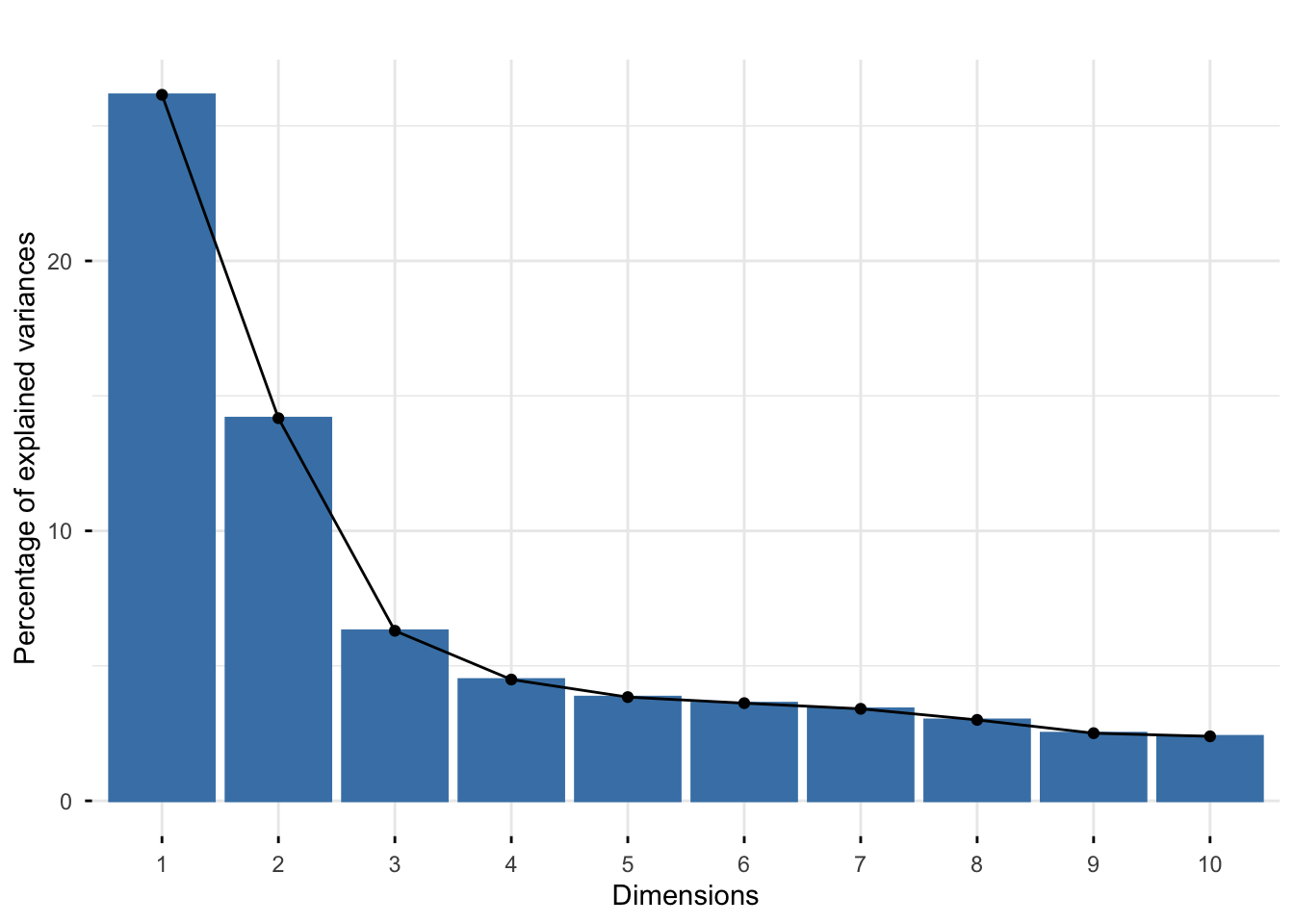
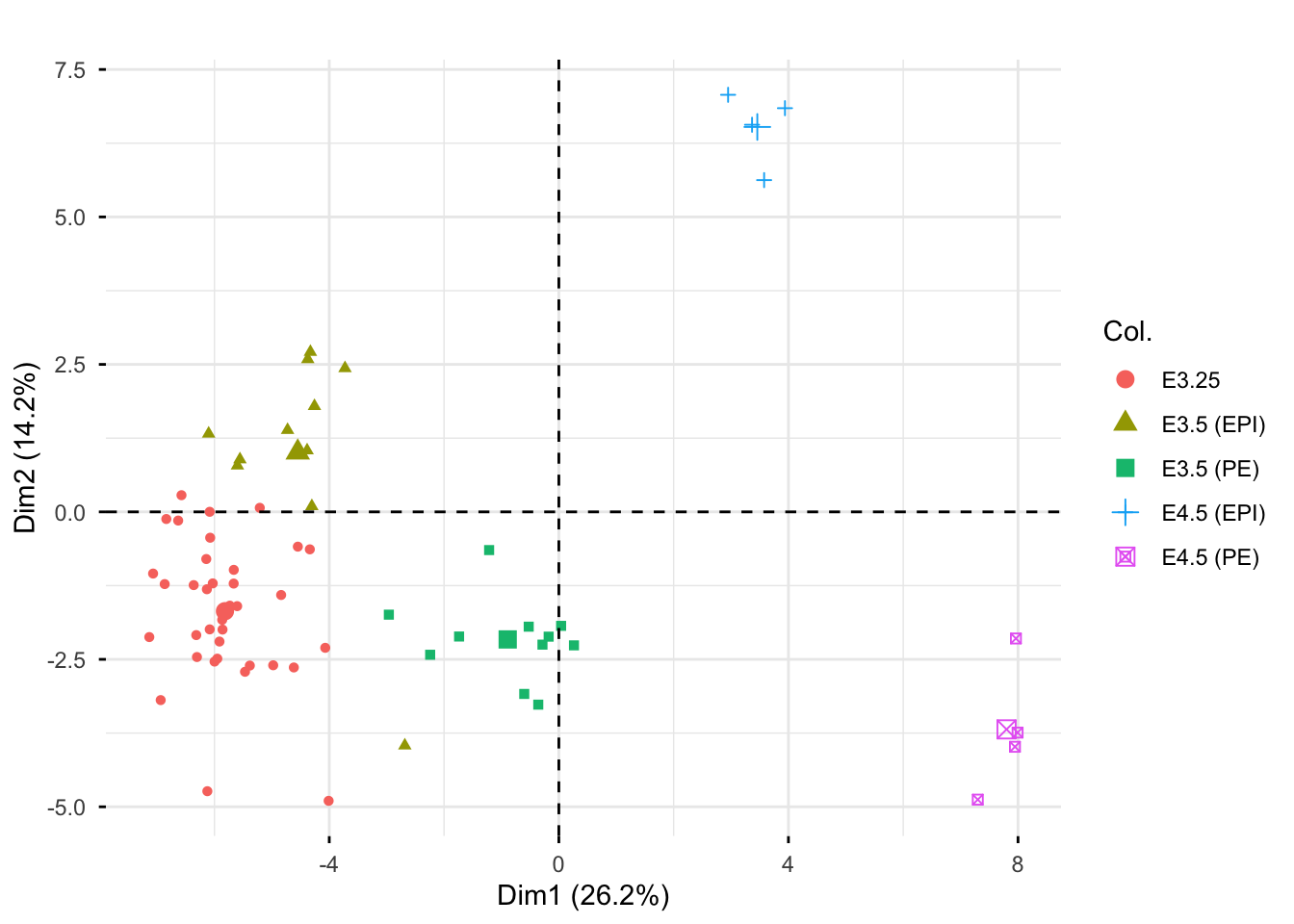
Rows: 25
Columns: 9
$ RedMeat <dbl> 10.1, 8.9, 13.5, 7.8, 9.7, 10.6, 8.4, 9.5, 18.0, 10.2, 5.3, …
$ WhiteMeat <dbl> 1.4, 14.0, 9.3, 6.0, 11.4, 10.8, 11.6, 4.9, 9.9, 3.0, 12.4, …
$ Eggs <dbl> 0.5, 4.3, 4.1, 1.6, 2.8, 3.7, 3.7, 2.7, 3.3, 2.8, 2.9, 4.7, …
$ Milk <dbl> 8.9, 19.9, 17.5, 8.3, 12.5, 25.0, 11.1, 33.7, 19.5, 17.6, 9.…
$ Fish <dbl> 0.2, 2.1, 4.5, 1.2, 2.0, 9.9, 5.4, 5.8, 5.7, 5.9, 0.3, 2.2, …
$ Cereals <dbl> 42.3, 28.0, 26.6, 56.7, 34.3, 21.9, 24.6, 26.3, 28.1, 41.7, …
$ Starch <dbl> 0.6, 3.6, 5.7, 1.1, 5.0, 4.8, 6.5, 5.1, 4.8, 2.2, 4.0, 6.2, …
$ Nuts <dbl> 5.5, 1.3, 2.1, 3.7, 1.1, 0.7, 0.8, 1.0, 2.4, 7.8, 5.4, 1.6, …
$ FrVeg <dbl> 1.7, 4.3, 4.0, 4.2, 4.0, 2.4, 3.6, 1.4, 6.5, 6.5, 4.2, 2.9, …