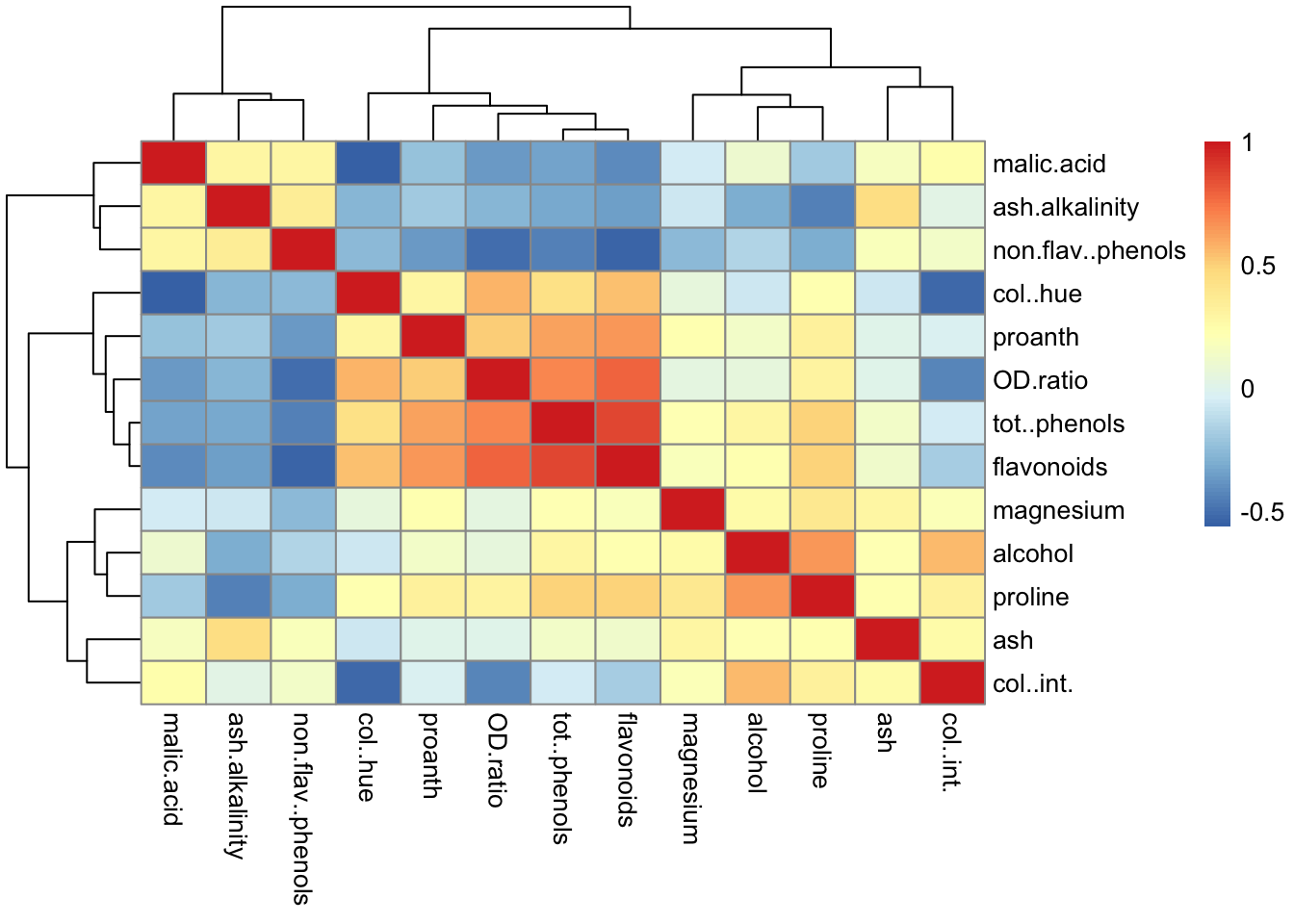
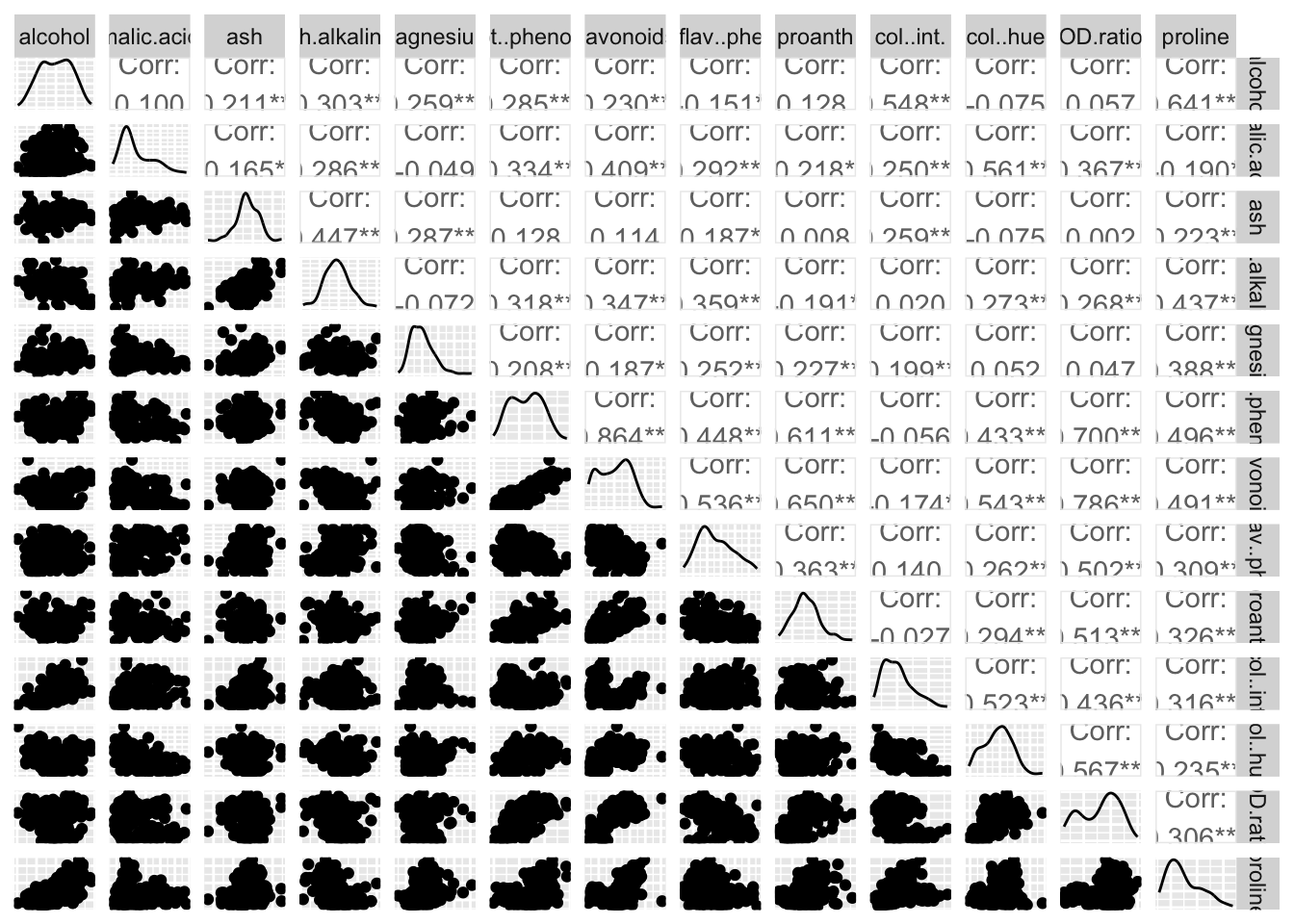
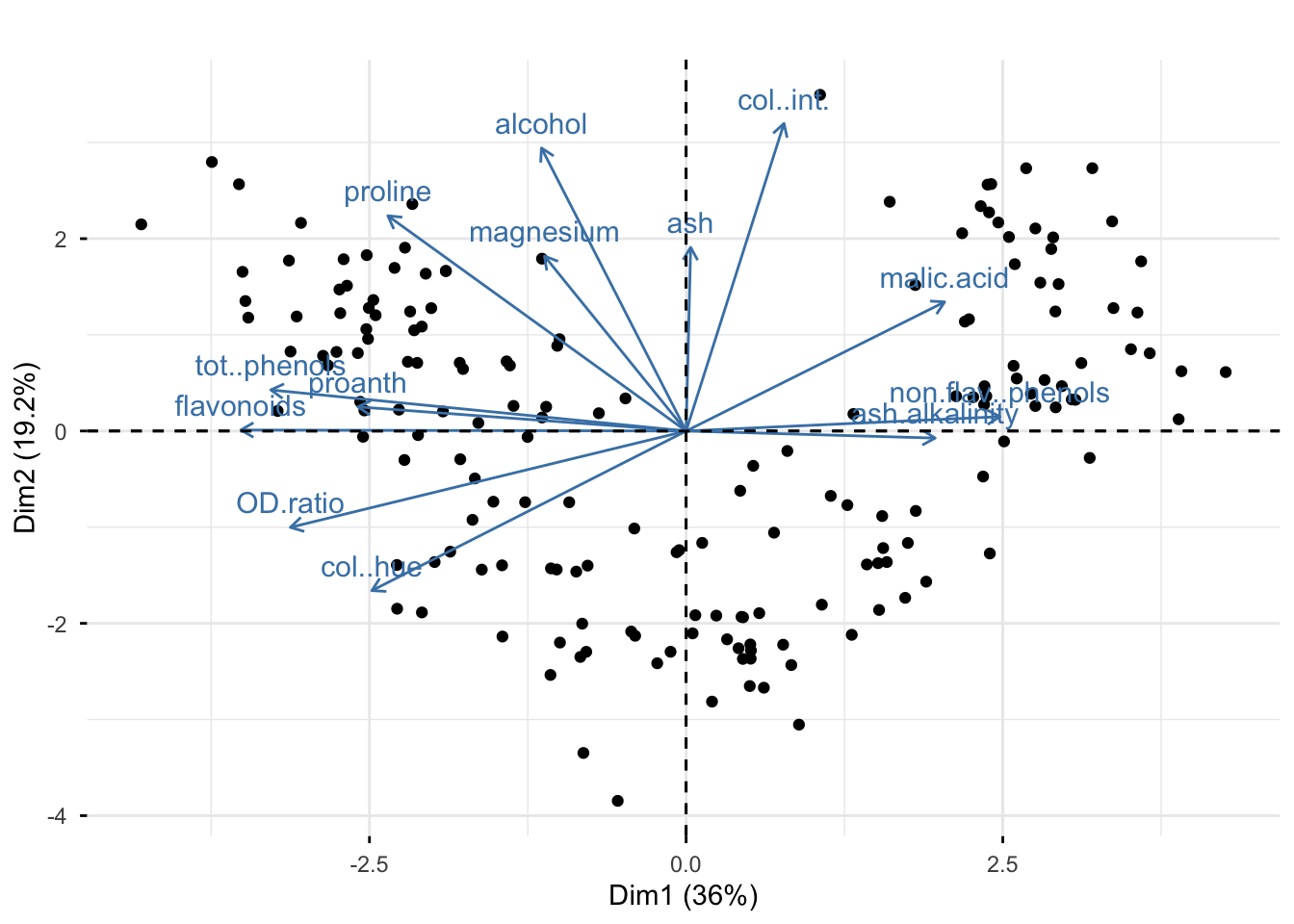
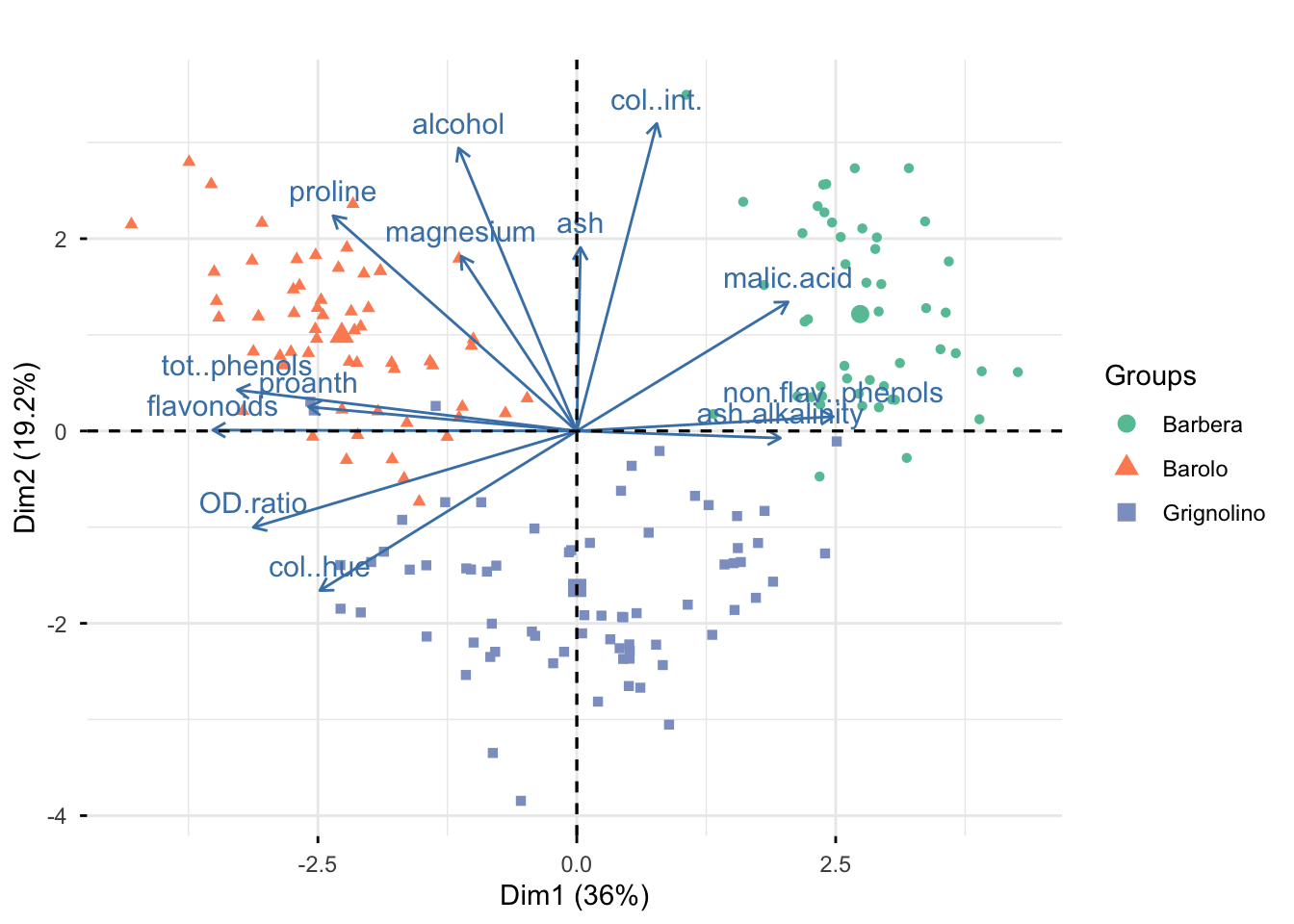
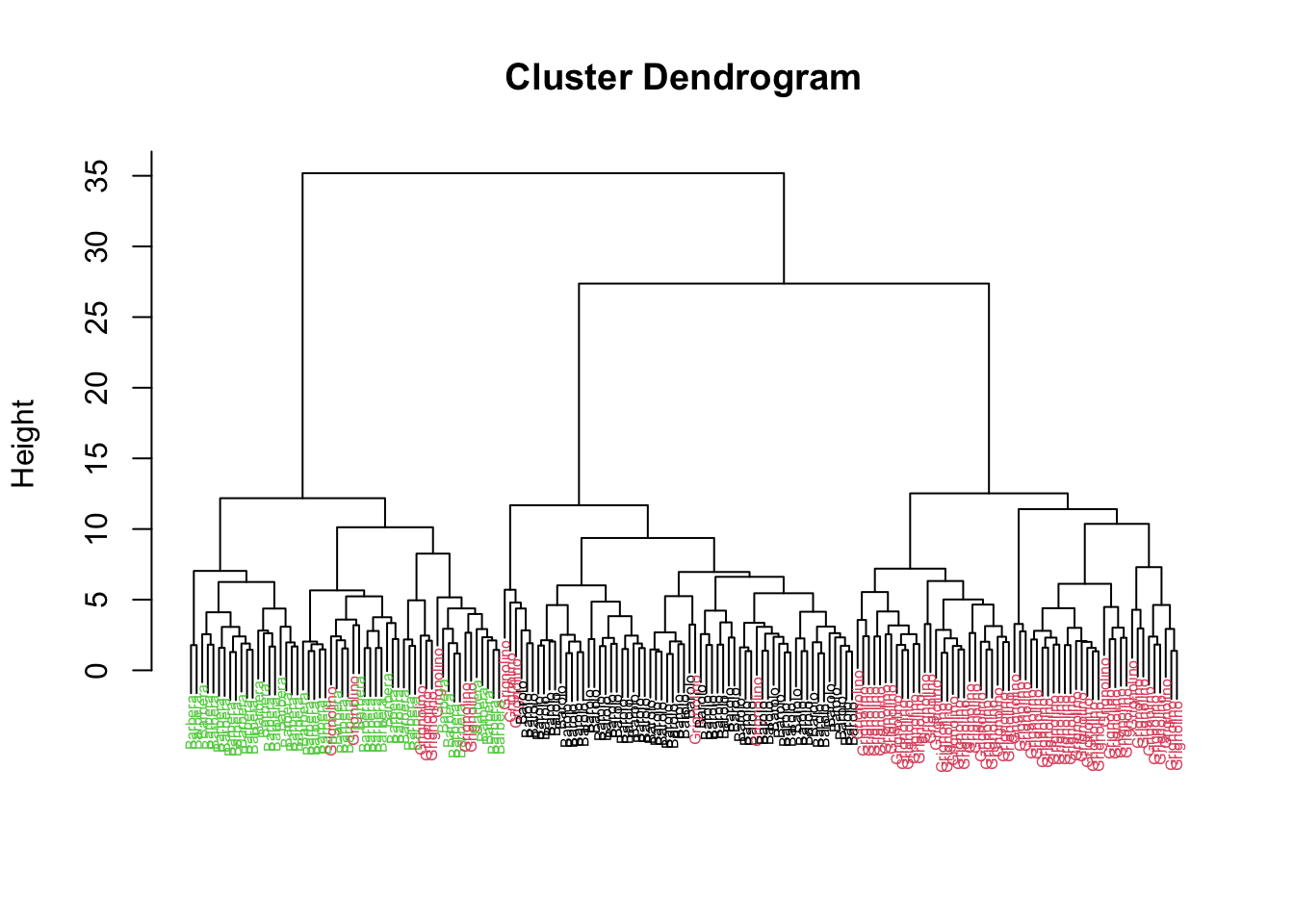
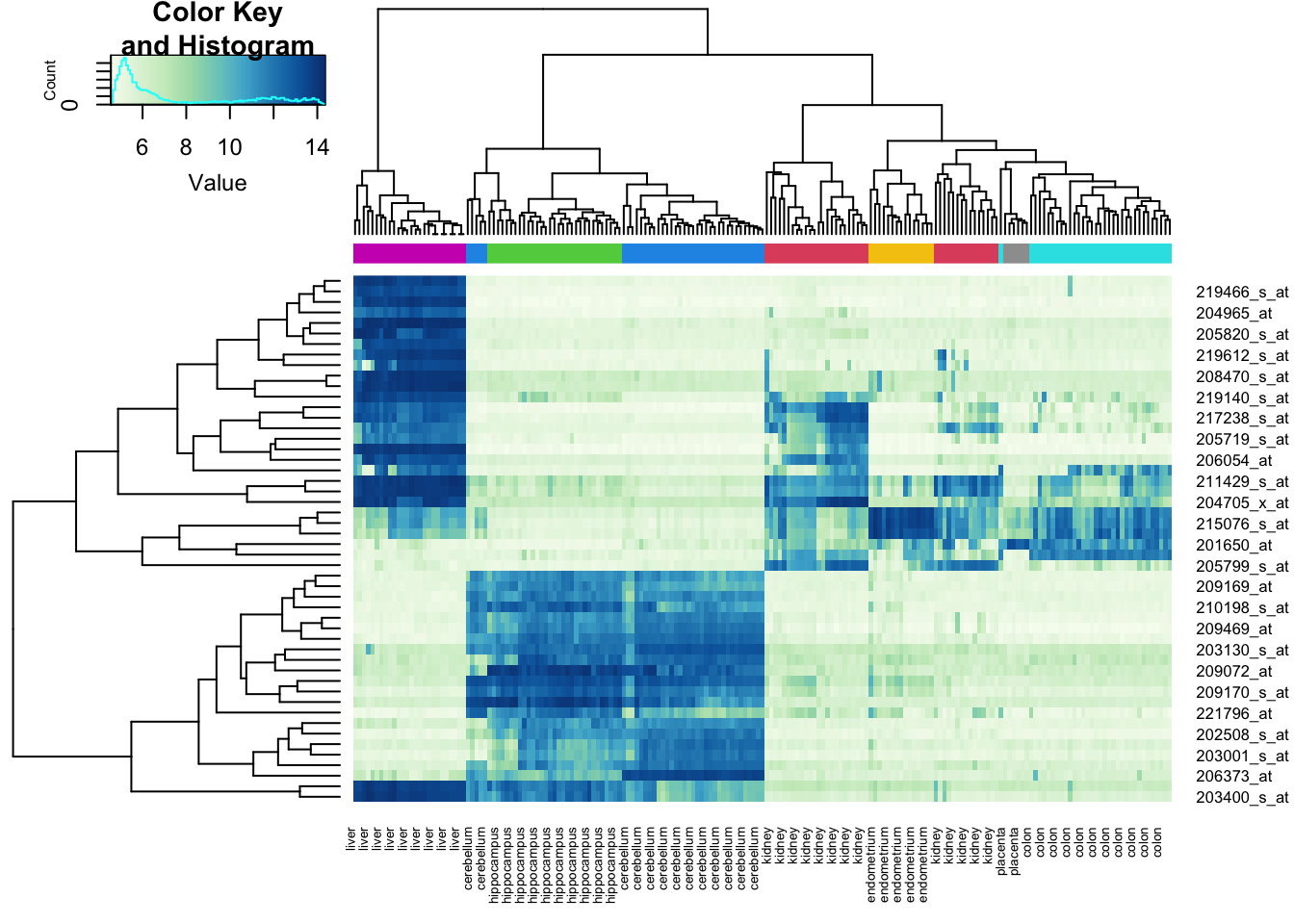
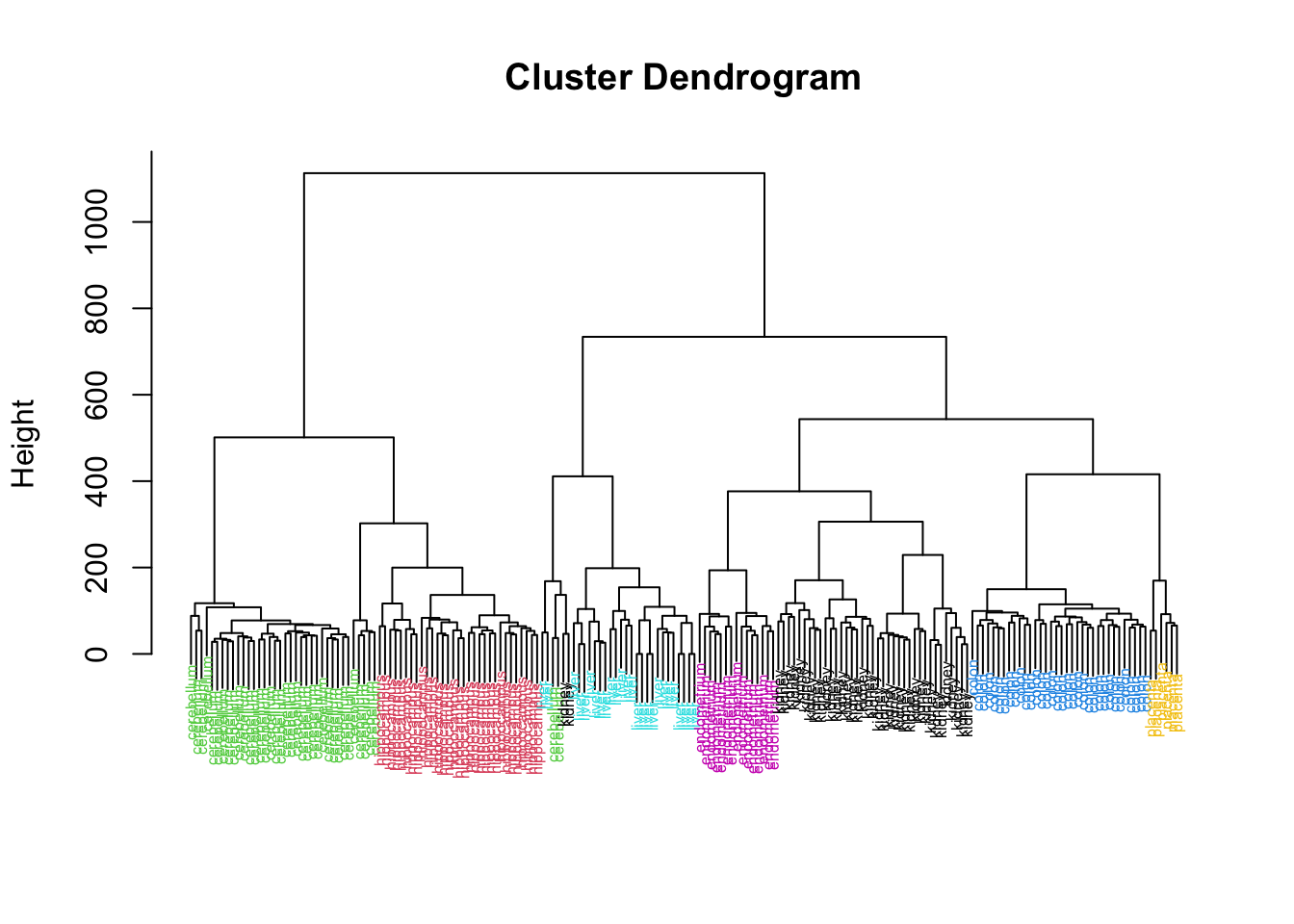
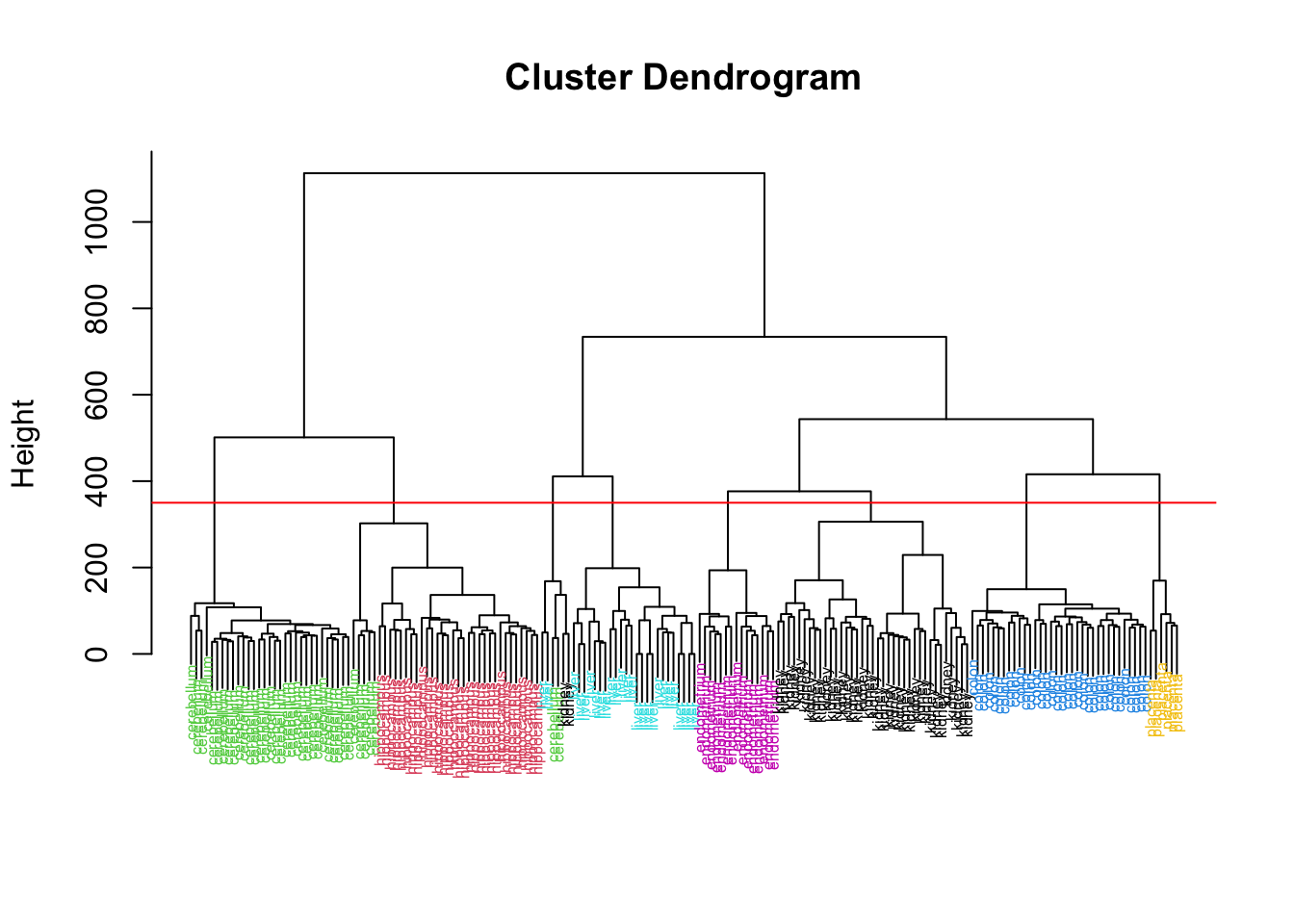
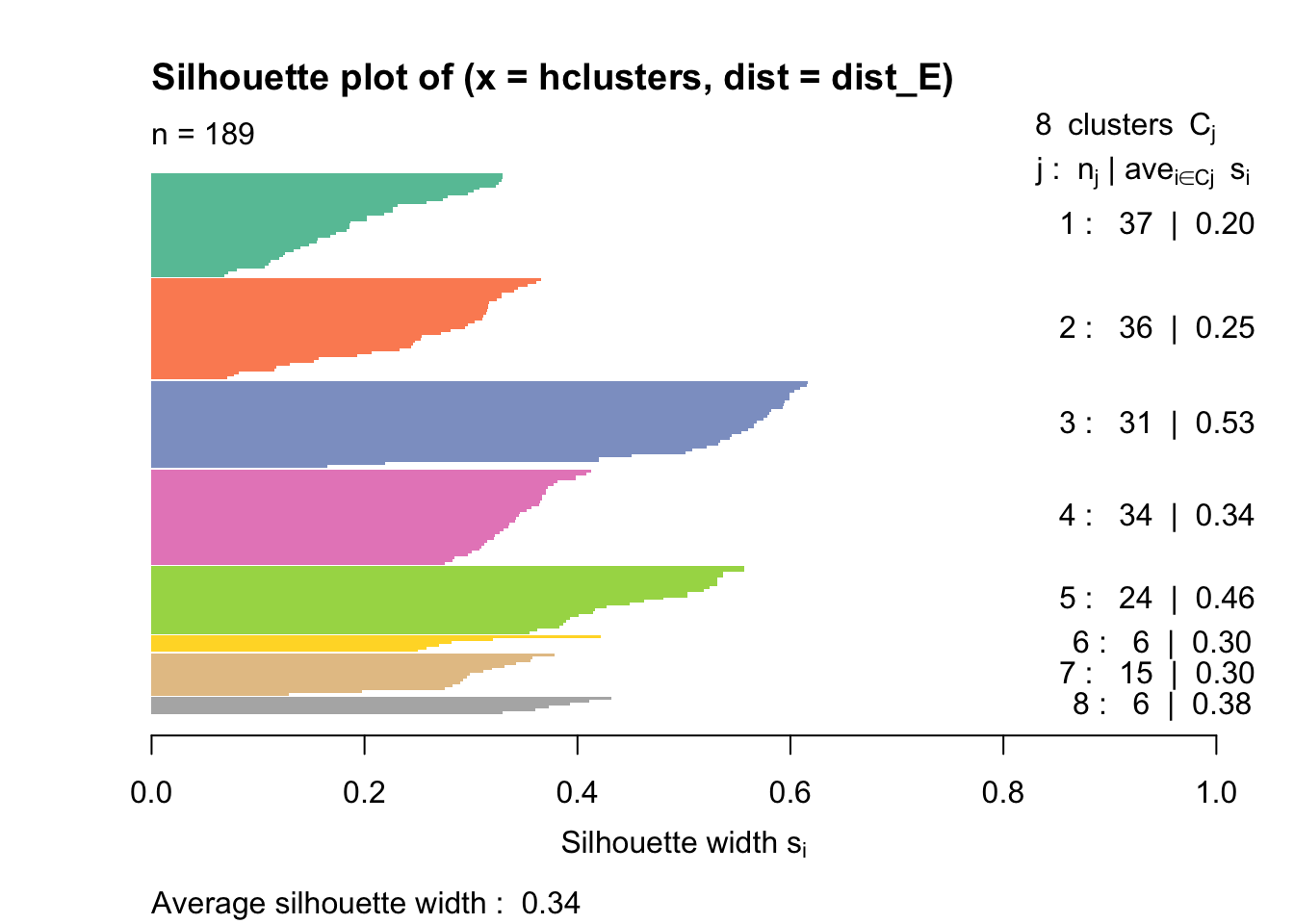
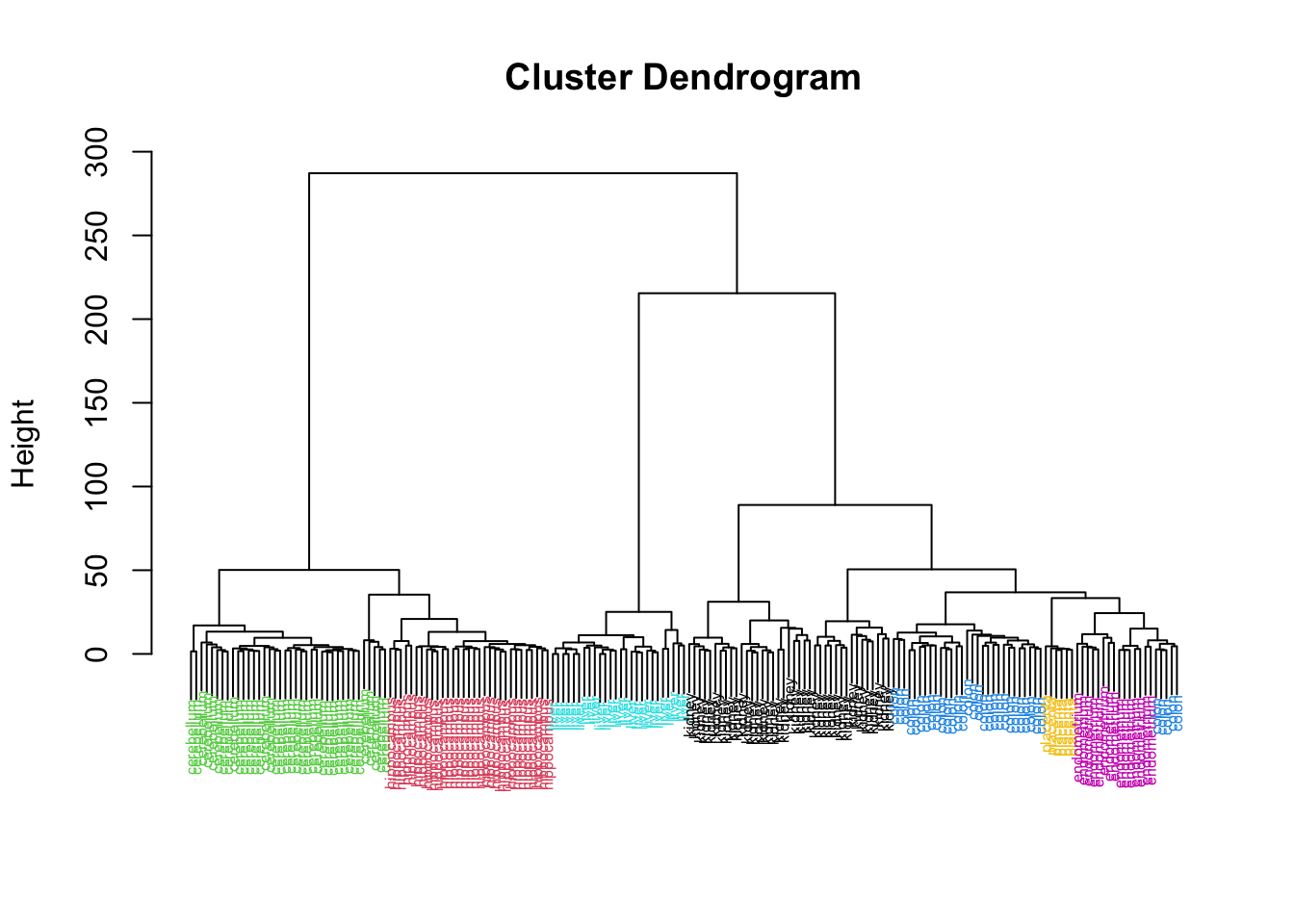
Rows: 177
Columns: 14
$ vintages <fct> Barolo, Barolo, Barolo, Barolo, Barolo, Barolo, Baro…
$ alcohol <dbl> 13.20, 13.16, 14.37, 13.24, 14.20, 14.39, 14.06, 14.…
$ malic.acid <dbl> 1.78, 2.36, 1.95, 2.59, 1.76, 1.87, 2.15, 1.64, 1.35…
$ ash <dbl> 2.14, 2.67, 2.50, 2.87, 2.45, 2.45, 2.61, 2.17, 2.27…
$ ash.alkalinity <dbl> 11.2, 18.6, 16.8, 21.0, 15.2, 14.6, 17.6, 14.0, 16.0…
$ magnesium <dbl> 100, 101, 113, 118, 112, 96, 121, 97, 98, 105, 95, 8…
$ tot..phenols <dbl> 2.65, 2.80, 3.85, 2.80, 3.27, 2.50, 2.60, 2.80, 2.98…
$ flavonoids <dbl> 2.76, 3.24, 3.49, 2.69, 3.39, 2.52, 2.51, 2.98, 3.15…
$ non.flav..phenols <dbl> 0.26, 0.30, 0.24, 0.39, 0.34, 0.30, 0.31, 0.29, 0.22…
$ proanth <dbl> 1.28, 2.81, 2.18, 1.82, 1.97, 1.98, 1.25, 1.98, 1.85…
$ col..int. <dbl> 4.38, 5.68, 7.80, 4.32, 6.75, 5.25, 5.05, 5.20, 7.22…
$ col..hue <dbl> 1.05, 1.03, 0.86, 1.04, 1.05, 1.02, 1.06, 1.08, 1.01…
$ OD.ratio <dbl> 3.40, 3.17, 3.45, 2.93, 2.85, 3.58, 3.58, 2.85, 3.55…
$ proline <dbl> 1050, 1185, 1480, 735, 1450, 1290, 1295, 1045, 1045,…